Final Diagnosis and a Discussion of the Differentials:
On the H&E-stained sections, the lymph node has architectural effacement by nodular and diffuse lymphoid proliferations. Nodular areas have mostly small B-lymphocytes and histiocytes, while the diffuse areas have abundant small T-lymphocytes and histiocytes. Scattered atypical large cells with morphologic and immunophenotypic features of lymphocyte predominant (LP) or popcorn cells (Figure 1 insets) are admixed in both types of areas, and they are more numerous in the diffuse areas, however, no confluent sheets of these cells are present. LP cells have markedly irregular large nuclei, prominent distinct nucleoli and are positive for CD20. These are also positive for BCL6, CD10 (focal, weak), CD45, CD79a, OCT2 and PAX5, and negative for CD30 and MUM1 (not shown). The nodular areas have expanded follicular dendritic cell (FDC) meshworks (CD21+) while the diffuse areas have a loss of the FDCs. CD3 and PD1 highlight numerous background T-cells, which also form rosettes around the LP cells. This is a “Nodular lymphocytic predominant Hodgkin lymphoma (NLPHL),” with patterns A and E (WHO 2022) based on the morphologic and immunophenotypic features.
NLPHL is a germinal center-derived B-cell neoplasm composed of scattered large neoplastic B-cells with multi-lobated nuclei (LP cells) within nodules of mantle zone B-cells and FDCs. In the variant histological growth patterns small B-cells are few and/or nodules are infrequent.
NLPHL is rare (incidence 0.11/100,000 person years) with higher incidences reported in the US, Europe and Middle East compared to Asia. It has a wide age distribution (childhood to >80 years, average 30-60 years) and a male:female ratio of 2.5-3:1. NLPHL usually involves lymph nodes, cervical or axillary regions frequently (40%) and iliac or inguinal regions occasionally (20%). Most patients (60%) present in an early stage with painless, localized lymphadenopathy, sometimes present for years. About 40% present in advanced stages with bulky disease. B-symptoms (fever, night sweats and weight loss) are not common, but they can be the presenting symptoms in 15-20% patients. The etiology of NLPHL is unknown. Rarely, genetic predispositions (familial cases with germline NPAT mutations or TET2 haploinsufficiency), primary immunodeficiency syndromes (Hermansky-Pudlak syndrome type 2) and autoimmune lymphoproliferative syndrome have been associated with NLPHL. In contrast to Classic Hodgkin Lymphoma (CHL), EBV positivity is rare. The pathogenesis involves genomic alterations of the LP cells and their immunological interactions with the microenvironment. LP cells are clonal germinal center B-cells with evidence of ongoing somatic hypermutations and intraclonal diversity in their rearranged IG genes. BCL6 translocations (46-48%) or amplifications (27%) are common. It is postulated that preceding bacterial infections lead to an extensive immune reaction in susceptible patients. Moraxella-derived antigens have been reported to play an additive role in the Ig-D positive cases. The close interaction between the rosetting PD1+ T-follicular helper (TFH) cells and the LP cells via immunological synapses suggests a role for the microenvironment in lymphomagenesis.
Macroscopically, the involved lymph nodes are enlarged and firm with a grey-tan and often nodular cut surface. Histologically, there is a partial or complete architectural effacement by a nodular proliferation of small lymphoid cells, sometimes with diffuse areas. Scattered large neoplastic LP cells are present with multi-lobated nuclei (popcorn-like), one or more nucleoli, and pale cytoplasm. The background has many non-neoplastic histiocytes, B-cells, and T-cells, including a subset of PD1+ TFH cells that form rosettes around the LP cells. Neutrophils and eosinophils are absent. The nodular areas are associated with FDC meshworks (CD21+, CD23+). Sclerosis may be seen, especially in recurrent cases. On immunohistochemistry, the LP cells stain positive for pan-leukocyte markers (CD45), B-cell markers (CD20, CD79a, CD22), B-cell transcription factors (PAX5, OCT2, BOB1), and germinal center markers (BCL6, HGAL, LMO2; while CD10 is usually negative). CD15, CD30, and EBV (EBER) are negative. Ig-D expression can be observed in cases associated with Moraxella sp. infection. Background TFH cells are abundant and are positive for CD3, CD4, PD1, ICOS, CD57 and CXCL13. Six growth patterns (A-F) have been described based on the histologic and immunophenotypic characteristics (Figure 3). “Typical B-cell rich” patterns (patterns A and B) are more common (75%) than the “variant patterns” (patterns C-F) (25%). In the variant patterns small B-cells are few and/or nodules are infrequent. More than one pattern may be present in the same patient. The identification of at least a focal nodular architecture is essential to render the diagnosis of NLPHL, especially in the diffuse T-cell/histiocyte rich large B-cell lymphoma-like (THRLBCL) pattern.
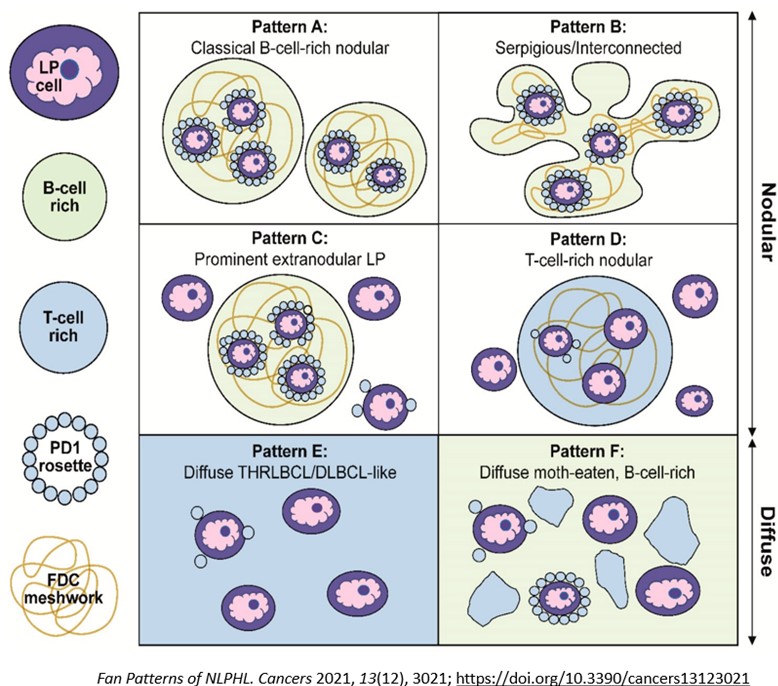
Figure 3
NLPHL can transform into a large B-cell lymphoma which can have heterogeneous appearances. On transformation, there is loss of nodular architecture and FDC meshworks, and there are decreased non-neoplastic B-cells accompanied by increased T-cells, histiocytes, and a variable proportion of increased LP cells. Sometimes NLPHL, diffuse large B-cell lymphoma and THRLBCL-like areas occur in the same lymph node. NLPHL with variant patterns, particularly pattern E (THRLBCL-like) is frequently observed when liver, spleen, and bone marrow are involved, and it must be distinguished from transformation to a large B-cell lymphoma. Ample tissue sampling is critical since distinction between NLPHL with an extensive pattern E component and a THRLBCL may not be possible on core biopsies or small tissue samples. Infra-diaphragmatic and splenic involvement as well as clonal IG rearrangement detected at initial diagnosis are risk factors for transformation to a large B-cell lymphoma.
NLPHL usually has an indolent clinical course with a 10-year overall survival >90% and a progression-free survival >75%. Relapses are more frequent compared to CHL. Variant growth patterns, male sex, and low serum albumin level are risk factors for relapse. Variable therapy protocols including excision, immunotherapy, chemotherapy, and radiation therapy are used based on the patient's age and disease stage. In patients with a limited stage disease, watchful waiting after resection in pediatric patients and active surveillance after radiation or chemotherapy in adults is used. Patients with advanced stage disease require systemic therapy usually combined with Rituximab. Even in the case of relapse, most patients do not require high-dose chemotherapy or autologous stem cell transplantation and can usually be treated sufficiently with low-intensity approaches such as a single-agent anti-CD20 antibody treatment.
The International Consensus Classification (ICC) (2022) adopted a new terminology for NLPHL, “nodular lymphocyte predominant B-cell lymphoma (NLPBL)” since there are biological and clinical similarities of NLPHL to indolent B-cell non-Hodgkin lymphoma (NHL) and major differences between NLPHL and CHL, justifying it to be a type of B-cell lymphoma. Also, standard treatments of NLPHL and CHL are different, which further supports adoption of the new terminology. B-NHL directed treatment strategies are more commonly used in NLPHL. Like WHO, ICC recognizes the importance of recognizing variant histologic patterns in NLPBL, wherein Fan patterns A, B, and C are combined into “typical” or grade 1, and Fan patterns D, E, and F into “variant” or grade 2 patterns. Grade 2 histology may warrant treatment for DLBCL, but clinical features should play a role in treatment decisions.
T-cell/histiocyte rich large B-cell lymphoma (THRLBCL)
Lymph nodes show a diffuse growth of lymphocytes (predominantly T-cells) and histiocytes
with scattered large neoplastic B-cells with large pleomorphic nuclei and variably
prominent nucleoli. Similar to NLPHL, the immunophenotype includes positivity for
B-cell markers but TFH rosettes and FDC meshworks are absent (negative CD21 and CD23).
Most importantly, no nodular proliferation is seen. THRLBL-like pattern and transformation
to THRLBL are in the spectrum of NLPHL, but de novo THRLBL is associated with a more
aggressive clinical course, and its distinction, although challenging, can be important.
Lymphocyte-rich Classic Hodgkin Lymphoma (LRCHL)
Lymph node architecture may be similar to NLPHL with nodular and sometimes diffuse
proliferation of lymphocytes composed of mantle-zone B-cells, and no neutrophils and
eosinophils. The scattered, neoplastic Hodgkin/Reed-Sternberg (HRS) cells have bi-
to multi-nucleation with prominent eosinophilic nucleoli and pale cytoplasm. On H&E,
these cells can be indistinguishable from the LP cells and immunohistochemistry is
essential for distinction. HRS cells are positive for CD15 and CD30, have minimal
expression of B-cell markers, and are negative for CD45. Rarely, LP cells of NLPHL
can express CD15 or CD30, but their lack of nuclear expression of STAT6 can help in
distinguishing the two.
Nodal T-follicular helper (TFH) cell lymphoma, angioimmunoblastic-type (nTFHL-AI)
This is a subtype of T-cell lymphoma that clinically presents with systemic disease,
bulky lymphadenopathy and cutaneous manifestations. Lymph nodes show a diffuse architectural
effacement by a polymorphous T-cell lymphoid infiltrate in a background of prominent
vasculature and a mixed inflammatory background including eosinophils. The neoplastic
T-cells are small- to intermediate-sized with a clear cytoplasm and moderate cytologic
atypia, and are positive for TFH markers (CD3, CD4, PD1). A subset of cases can have
HRS-like large cells that are positive for CD30.
References:
1. WHO Classification of Tumours Editorial Board. Haematolymphoid tumours [Internet;
beta version ahead of print]. Lyon (France): International Agency for Research on
Cancer; 2022 [cited 08/11/2023]. (WHO classification of Tumours Series, 5th ed.; vol.
11). Available from: https://tumourclassification.iarc.who.int/chapters/63.
2. Elias Campo, Elaine S. Jaffe, James R. Cook, Leticia Quintanilla-Martinez et al;
The International Consensus Classification of Mature Lymphoid Neoplasms: a report
from the Clinical Advisory Committee. Blood 2022; 140 (11): 1229–1253. doi: https://doi.org/10.1182/blood.2022015851
3. Younes S, Rojansky RB, Menke JR, Gratzinger D, Natkunam Y. Pitfalls in the Diagnosis
of Nodular Lymphocyte Predominant Hodgkin Lymphoma: Variant Patterns, Borderlines
and Mimics. Cancers. 2021; 13(12):3021. https://doi.org/10.3390/cancers13123021
4. Fan Z, Natkunam Y, Bair E, Tibshirani R, Warnke RA. Characterization of Variant
Patterns of Nodular Lymphocyte Predominant Hodgkin Lymphoma with Immunohistologic
and Clinical Correlation. Am J Surg Pathol. 2003 Oct;27(10):1346-56. doi: 10.1097/00000478-200310000-00007.
PMID: 14508396.
5. Eichenauer DA, Fuchs M. Treatment of Nodular Lymphocyte-Predominant Hodgkin Lymphoma:
Where Do We Stand? Where Do We Go? Cancers. 2023; 15(13):3310. https://doi.org/10.3390/cancers15133310
6. Hartmann, S., Dojcinov, S., Dotlic, S. et al. The spectrum of nodular lymphocyte
predominant Hodgkin lymphoma: a report of the lymphoma workshop of the 20th meeting
of the European Association for Haematopathology. Virchows Arch (2023). https://doi.org/10.1007/s00428-023-03554-1
7. Tousseyn, T.A., R.L. King, F. Fend, A.L. Feldman, P. Brousset and E.S. Jaffe (2023).
"Evolution in the definition and diagnosis of the Hodgkin lymphomas and related entities."
Virchows Arch 482(1): 207-226.
Board type review questions:
1. The following statements are true regarding Nodular Lymphocytic Predominant Hodgkin
lymphoma except:
A. There is a male predominance and an indolent clinical course.
B. Neoplastic B-cells have an irregular, multi-lobated nucleus with prominent nucleoli
and pale cytoplasm.
C. It is a germinal center derived B-cell lymphoma.
D. Fan patterns C, D, E, and F are considered as “typical” patterns.
2. LP cells in Nodular Lymphocytic Predominant Hodgkin lymphoma have the following
immunophenotype:
A. BCL6-, CD15+, CD20-, CD30+, CD79a-, EBV+, PAX5+, STAT6+ (nuclear)
B. BCL6+, CD3+, CD4+, CD5+, CD7-, CD10+, CD15-, CD20-, CD30+, EBV+, PD1+
C. BCL6+, CD15-, CD20+, CD30-, CD45+, CD79a+, EBV-, PAX5+
D. BCL6-/+, CD10-, CD15-/+, CD20+, CD30+, CD45+, CD79a+, EBV+, MUM1+
C. LP cells are positive for B-cell markers like CD20, CD79a, PAX5, for some germinal center markers (BCL6) and for the pan-leukocyte marker CD45. These are negative for CD15, CD30 and EBV.
Immunoprofile of other choices are for the following entities:
A. LRCHL
B. nTFHL-AI
D: EBV-positive diffuse large B-cell lymphoma (not discussed). This is a large B-cell lymphoma in which majority of the cells harbor EBV; there is no known history of a prior lymphoma or an underlying immune deficiency/dysregulation (other than immune senescence).